1. 引言
α-一卤甲基酮和α,α-二卤甲基酮作为重要的有机合成中间体,在各类有机物的合成中被广泛使用 [1] - [6]。例如它们常被用于环丙烷 [7]、酰胺 [8]、醛 [9]、炔 [10]、酮 [11]、α,β-二卤甲基酮 [12] [13] [14] [15]、不饱和酮或1,4-二酮、炔醇、杂环化合物等的合成,除此之外,α-一卤甲基酮和α,α-二卤甲基酮骨架也广泛存在于天然产物、药物、农药中,并表现出较优的生物活性,比如抗真菌、抗艾滋病毒和抗肿瘤活性等 [16] (图1)。鉴于α-一卤甲基酮和α,α-二卤甲基酮类化合物的重要作用,建立该类化合物的合成新方法具有重要的意义。
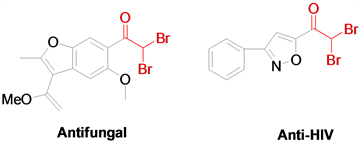
Figure 1. Important α,α-dihalomethyl ketone compounds
图1. 重要的α,α-二卤甲基酮化合物
到目前为止,人们对α-一卤甲基酮和α,α-二卤甲基酮的合成方法也进行了研究,除利用苯乙酮与溴源的溴化反应外 [17],还有以下三种常用的方法:一是以炔烃为起始原料(图2(I)) [18] [19] [20] [21],PhI(OAc)2、氧代酮、氯化铁、
为催化剂或氧化剂的溴代反应。二是使用单质溴作为溴源,实现了α-一卤甲基酮和α,α-二卤甲基酮的高效合成(图2(II)) [22] [23],但该法较难停留在α-一卤甲基酮的合成。三是使用1,3-二氯-5,5-二甲基乙内酰脲(DCDMH)为选择性氯代试剂来制备α-单氯酮或α,α-二氯酮(图2(III)) [24] [25]。

Figure 2. Direct halogenation synthesis of α-mono- and α,α-dihalo-methylketones
图2. 直接卤代法合成α-一卤甲基酮和α,α-二卤甲基酮
与上述直接卤代方式制备α-一卤甲基酮和α,α-二卤甲基酮相比,α-多卤代酮的选择性脱卤也是较为有效的方式,例如在金属钌、铟、碲或离子液体等催化下的选择性脱卤反应(图3(I)) [26] [27] [28] [29],这些反应大多数在选择性方面受到限制,因此建立高选择性、便捷的脱卤反应显得非常重要,目前阿布都热西提课题组在2020年提出了以H2O-HBr为还原条件,对α,α,α-三溴甲基酮进行脱溴,实现高选择性地合成α-一溴甲基酮和α,α-二溴甲基酮(图3(II)) [16]。

Figure 3. Selective debromination of α-polyhalogenones
图3. α-多卤代酮的选择性脱溴反应
受上述反应鼓舞,本课题组继续研究α-多溴代甲基酮的选择性脱溴反应,在本论文中,从α,α,α-三溴甲基酮出发,通过改变反应温度和溴水质量、溶剂等因素,实现了α-多溴代甲基酮的高效选择性控制还原,分别获得α-一溴甲基酮和α,α-二溴甲基酮等化合物。该类反应产物易于调控,产率较优,底物适用范围广,官能团耐受力强,并对反应机理进行了研究,初步推测了溴水催化下的α-多溴代甲基酮类化合物的选择性脱溴反应机理。
2. 实验部分
2.1. 仪器和试剂
HMS-14数字型加热磁力搅拌器(上海泰坦科技股份有限公司);IKA-HB10型旋转蒸发仪(上海兴创仪器设备有限公司);XT4-100B型熔点仪(北京市科仪电光仪器厂);Varian Inoova-400型核磁共振谱仪(美国Varian公司);研究过程中使用的所有试剂均在泰坦科技探索平台购买。
2.2. 实验方法
2.2.1. 化合物2的合成
在15 mL厚壁耐压瓶中依次加入磁子、0.1 mmol 1、1,4-二氧六环为溶剂,添加20 mg溴水溶液(Br2(3.2 mg)、水(16.8 mg)),80℃下进行反应,底物全部反应完后,用薄层层析板纯化,石油醚:二氯甲烷(3:1体积比),得到产物2a~2o。因为都是已知物,所以只提供了核磁共振氢谱。
2.2.2. 化合物3的合成
在15 mL厚壁耐压瓶中依次加入磁子、0.1 mmol 1、1,4-二氧六环为溶剂,添加30 mg溴水溶液(Br2(4.8 mg)、水(25.2 mg)),120℃下进行反应,底物全部反应完后,用薄层层析板纯化,石油醚:二氯甲烷(3:1体积比),得到产物3a~3o。因为都是已知物,提供了核磁共振氢谱。
2.3. 化合物的结构与表征
2,2-二溴-1-苯基乙酮(2a) [8]:23.9 mg,白色固体,分离产率86%。m.p. 36.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.05 (t, J = 4.0 Hz, 2H),7.63 (t, J = 8.0 Hz, 1H),7.50 (t, J = 8.0 Hz, 2H),6.70 (s, 1H)。
2,2-二溴-1-(2-氯苯基)-乙酮(2b):27.9 mg,无色液体,分离产率90%。1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.62 (d, J = 8.0 Hz, 1H),7.50~7.46 (m, 2H),7.40~7.34 (m, 1H),6.76 (s, 1H)。
2,2-二溴-1-(3-氯苯基)-乙酮(2c):20.2 mg,无色液体,分离产率65%。1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.05 (d, J = 4.0 Hz, 1H),7.98 (d, J = 8.0 Hz, 1H),7.60 (t, J = 4.0 Hz, 1H),7.45 (t, J = 8.0 Hz, 1H),6.60 (s, 1H)。
2,2-二溴-1-(4-氯苯基)-乙酮(2d) [8]:24.2 mg,白色固体,分离产率78%。m.p. 91.0℃~92.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.07 (d, J = 12 Hz, 2H),7.50 (d, J = 8.0 Hz, 2H),6.60 (s, 1H)。
2,2-二溴-1-(4-溴-苯基)-乙酮(2e) [8]:28 mg,白色固体,分离产率78%。m.p. 90.0℃~91.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.98 (d, J = 8.0 Hz, 2H),7.67 (d, J = 8.0 Hz, 2H),6.60 (s, 1H)。
2,2-二溴-1-(4-氟苯基)-乙酮(2f) [8]:21.8 mg,白色固体,分离产率74%。m.p. 36℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.17~8.14 (m, 2H),7.21~7.17 (m, 2H),6.62 (s, 1H)。
4-(2,2-二溴-乙酰基)-苄腈(2g) [8]:22.6 mg,白色固体,分离产率75%。m.p. 97.0℃~98.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.23 (d, J = 4.0 Hz, 2H),7.83 (d, J = 8.0 Hz, 2H),6.58 (s, 1H)。
4-(2,2-二溴-乙酰基)-苯甲酸甲酯(2h) [8]:25.7 mg,白色固体,分离产率77%。m.p. 41.0℃~42.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.13 (s, 4H),6.65 (s, 1H),3.95 (s, 3H)。
2,2-二溴-1-(4-溴-2-氟-苯基)-乙酮(2i) [8]:27.8 mg,白色固体,分离产率74%。m.p. 47.0℃~48.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.88 (t, J = 8.0 Hz, 1H),7.47 (t, J = 4.0 Hz, 1H),7.42 (d, J = 12.0 Hz, 1H),6.78 (s, 1H)。
2,2-二溴-1-对甲苯基乙酮(2j) [8]:20.5 mg,白色固体,分离产率70%。m.p. 95.0℃~96.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.98 (d, J = 8.0 Hz, 2H),7.30 (d, J = 8.0 Hz, 2H),6.68 (s, 1H),2.43 (s, 1H)。
2,2-二溴-1-(4-叔丁基-苯基)-乙酮(2k):25.8 mg,无色液体,分离产率77%。1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.03 (d, J = 8.0 Hz, 2H),7.52 (d, J = 8.0 Hz, 2H),6.68 (s, 1H),1.34 (s, 9H)。
2,2-二溴-1-(4-甲氧基-苯基)-乙酮(2l):22.4 mg,无色液体,分离产率73%。1H NMR (400 MHz, CDCl3) δ: 8.06 (d, J = 8 Hz, 2H),6.96 (d, J = 12 Hz, 2H),6.65 (s, 1H),3.88 (s, 1H)。
2,2-二溴-1-噻吩-2-基乙酮(2m):22.3 mg,无色液体,分离产率79%。1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.00 (t, J = 4.0 Hz, 1H),7.98 (t, J = 4.0 Hz, 1H),7.20 (t, J = 4.0 Hz, 1H),6.48 (s, 1H)。
2,2-二溴-1-萘-2-基乙酮(2n) [8]:27 mg,白色固体,分离产率82%。m.p. 97.0℃~98.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.63 (s, 1H),8.09 (d, J = 4.0 Hz, 1H),7.99 (d, J = 4.0 Hz, 1H),7.94~7.84 (m, 2H),7.65 (t, J = 4.0 Hz, 1H),7.59 (t, J = 4.0 Hz, 1H),6.86 (s, 1H)。
1,1-二溴-4-苯基-丁烷-2-酮(2o):12.9 mg,无色液体,分离产率42%。1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.30 (t, J = 4.0 Hz, 2H),7.23~7.20 (m, 3H),5.74 (s, 1H),3.24 (t, J = 8.0 Hz, 2H),2.98 (t, J = 8.0 Hz, 3H)。
2-溴-1-苯基乙酮(3a) [8]:15.3 mg,白色固体,分离产率77%。m.p. 45.0℃~46.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.00 (d, J = 8.0 Hz, 2H), 7.63~7.59 (m, 1H),7.50 (t, J = 8.0 Hz, 2H),4.46 (s, 2H)。
2-溴-1-(2-氯苯基)-乙酮(3b):16.7 mg,无色液体,分离产率72%。1H NMR(CDCl3, TMS, 400 MHz) (ppm) δ 7.68 (d, J = 8.0 Hz, 1H),7.45 (d, J = 4.0 Hz, 2H),7.39~7.35 (m, 1H),4.52 (s, 2H)。
2-溴-1-(3-氯苯基)-乙酮(3c) [8]:14 mg,白色固体,分离产率60%。m.p. 39.0℃~40.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.96 (d, J = 4.0 Hz, 1H),7.87 (d, J = 8.0 Hz, 1H),7.60~7.57 (m, 1H),7.45 (t, J = 4.0 Hz, 1H),4.42 (s, 2H)。
2-溴-1-(4-氯苯基)-乙酮(3d) [8]:15.4 mg,白色固体,分离产率66%。m.p. 92.0℃~93.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.92 (d, J = 8.0 Hz, 2H),7.47 (d, J = 12.0 Hz, 2H),4.39 (s, 2H)。
2-溴-1-(4-溴苯基)-乙酮(3e) [8]:18.6 mg,白色固体,分离产率67%。m.p. 109.0℃~110.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.86 (d, J = 8.0 Hz, 2H),7.65 (d, J = 8.0 Hz, 2H),4.40 (s, 2H)。
2-溴-1-(4-氟苯基)-乙酮(3f) [8]:13.2 mg,白色固体,分离产率61%。m.p. 43.0℃~44.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.04~8.00 (m, 2H),7.20~7.14 (m, 2H),4.41 (s, 2H)。
4-(2-溴-乙酰基)-苄腈(3g) [8]:15.7 mg,白色固体,分离产率70%。m.p. 91.0℃~92.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.09 (d, J = 8.0 Hz, 2H),7.81 (d, J = 8.0 Hz, 2H),4.43 (s, 2H)。
4-(2-溴-乙酰基)-苯甲酸甲酯(3h) [8]:16.6 mg,白色固体,分离产率65%。m.p. 135.0℃~136.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.15 (d, J = 8.0 Hz, 2H),8.04 (d, J = 8.0 Hz, 2H),4.48 (s, 2H),3.95 (s, 3H)。
2-溴-1-(4-溴-2-氟苯基)-乙酮(3i):19.5 mg,无色液体,分离产率66%。1H NMR (400 MHz, CDCl3) δ: 7.81 (t, J = 8 Hz, 1H),7.39 (m, J = 8 Hz, 2H),4.46 (d, J = 4Hz, 2H)。
2-溴-1-对甲苯基乙酮(3j) [8]:14.5 mg,白色固体,分离产率68%。m.p. 49.0℃~50.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.90 (d, J = 8.0 Hz, 2H),7.30 (d, J = 8.0 Hz, 2H),4.43 (s, 2H),2.43 (s, 3H)。
2-溴-1-(4-叔丁基苯基)-乙酮(3k):18.2 mg,无色液体,分离产率71%。1H NMR(CDCl3, TMS, 400 MHz) (ppm) δ 7.93 (t, J = 4.0 Hz, 2H),7.52 (d, J = 8.0 Hz, 2H),4.44 (s, 2H),1.35 (s, 9H)。
2-溴-1-(4-甲氧基-苯基)-乙酮(3l):15.2 mg,无色液体,分离产率67%。1H NMR (400 MHz, CDCl3) δ: 7.95 (d, J = 8 Hz, 2H),6.94 (d, J = 8 Hz, 2H),4.36 (s, 1H),3.87 (s, 1H)。
2-溴-1-噻吩-2-基乙酮(3m):12.5 mg,无色液体,分离产率61%。1H NMR(CDCl3, TMS, 400 MHz) (ppm) δ 7.82 (s, 1H),7.73 (s, 1H),7.18 (s, 1H),4.37 (s, 2H)。
2-溴-1-萘-2-基乙酮(3n) [8]:19 mg,白色固体,分离产率76%。m.p. 81.0℃~82.0℃;1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 8.51 (s, 1H),8.04~7.97 (m, 2H),7.93~7.88 (m, 2H),7.65~7.56 (m, 2H),4.58 (s, 2H)。
1-溴-4-苯基丁烷-2-酮(3o):9.2 mg,无色液体,分离产率41%。1H NMR (CDCl3, TMS, 400 MHz) (ppm) δ 7.29 (t, J = 8.0 Hz, 2H),7.22~7.18 (m, 3H),3.84 (s, 2H),3.01~2.92 (m, 4H)。
3. 结果与讨论
3.1. α,α–二溴甲基酮类化合物的合成研究
3.1.1. 反应条件优化
α-多溴代甲基酮的高选择性控制脱溴反应仍然是一个巨大的挑战,在研究开始时,首先选择α,α,α-三溴苯乙酮(1a, 0.1 mmol)作为反应底物,在80℃下,加入100 mg溴水溶液(Br2(16 mg)、水(84 mg))对不同类型溶剂进行了筛选,令人欣喜的是,相较乙酸乙酯、乙醇、乙腈、四氢呋喃等溶剂(表1,Entries 2~5),以1,4-二氧六环作为溶剂时,目标产物α,α-二溴苯乙酮(2a)产率可达58% (表1,Entry 1)。因此在下一步条件优化时选用1,4-二氧六环为溶剂进行反应。紧接着,继续考查了溴水溶液质量对α-多溴代甲基酮的选择性脱溴反应的影响,研究表明溴水溶液质量也是实现选择性脱溴的关键所在,当使用20 mg溴水溶液,2a产率达到最优(表1,Entry 8),产率高达86%,增加或降低溴水质量均导致2a产率下降(表1,Entries 6~12)。最后,考察温度影响时发现,升高温度或降低温度均导致2a产率下降(表1,Entry 13),温度降低到60℃时,发现2a产率为73%,而温度升高至100℃时,2a的产率明显下降,为28%,并伴随58%产率的α-一溴苯乙酮3a生成。
通过上述反应条件筛选,确定了目标分子2a的最佳条件:α,α,α-三溴苯乙酮(1a,0.1 mmol)作为反应底物,在80℃的1,4-二氧六环溶剂中,溴水溶液20 mg (Br2(3.2 mg)、H2O (16.8 mg))为还原剂在3.5 h内可获得86%的2a。

Table 1. Optimization of reaction conditions for synthesis of α,α-dibromomethyl ketone compoundsa
表1. α,α-二溴甲基酮类化合物的合成反应条件优化a
aReaction conditions: 80℃, 1a (0.1 mmol), bromine water solution (Eq. (Br2):(Eq. H2O) = 0.2:9.3), solvent (1 mL).bIsolated yield.ctemperature was at 60℃.dtemperature was at 100℃.
3.1.2. 底物的扩展
在上述最优条件下,继续考察了该法的底物范围和官能团耐受性,结果如表2所示,研究结果表明:不同芳基取代的α,α,α-三溴甲基酮均能顺利地进行了脱溴反应,获得42%~90%产率的α,α-二溴甲基酮化合物(2a~2o),其中芳环上带有吸电子性质的取代基往往对2的产率有积极影响。考察芳环上取代基位置对产物2的产率影响时发现:与芳环上间位和对位有取代基相比,当羰基邻位上有取代基基时,脱溴反应更具优势(2b~2d),值得一提的是,缺电子芳香族和大共轭芳香族底物也可分别以79%和82%的产率顺利地实现选择性脱溴反应(2m~2n)。此外, α-多卤代烷基酮的脱溴反应同样能顺利进行,得到相应的产物2o,产率为42%,同时发现,该选择性脱溴反应在卤素、酯基、氰基、酮基等官能团存在下,同样也能实现2a~2o的有效合成。上述研究结果表明,该反应条件易于控制,底物适用范围广,官能团耐受性强,为α,α-二溴甲基酮化合物的制备提供了较优的合成策略。
Table 2. Synthesis of the α,α-dibromomethyl ketones 2a~2oa,b
表2. α,α-二溴甲基酮2a~2o的合成a,b
aReaction conditions: 80℃, 1a (0.1 mmol), bromine water solution (Eq. (Br2):(Eq. H2O) = 0.2:9.3), solvent (1 mL).bIsolated yield.
3.2. α-一溴甲基酮类化合物的合成研究
3.2.1. 反应条件优化
在α,α,α-三溴甲基酮化合物的单脱溴反应中发现,反应温度和溴水质量等对α,α-二溴甲基酮类化合物的合成均能产生较大影响,因此本实验中继续通过调整溴水质量和调控反应温度,以期实现α-一溴甲基酮类化合物的选择性合成。首先将反应温度升高至100℃,发现使用20 mg溴水时,就能获得58%产率的α-一溴苯乙酮3a,但继续增加溴水质量,3a的产率略微下降(表3,Entries 2~3)。因此改变反应温度,欣喜的是当反应温度提高到120℃,20 mg溴水存在下,3a产率有了明显提高,产率可达70% (表3,Entry 4),在此条件下,通过调整溴水质量,当使用30 mg溴水时3a产率达到最优,可至77% (表3,Entry 5)。但继续将温度提高到160℃时,3a产率明显下降,仅有25%产率的3a生成,并伴随大量全脱溴产物苯乙酮4a生成(表3,Entry 8)。
获得了3a的最优反应条件:1a (0.1 mmol)为原料,溴水溶液30 mg Br2(4.8 mg)为催化剂,H2O (25.2 mg)),温度为120℃,在1,4-二氧六环溶剂中进行时0.5 h内可获得77%的3a。
Table 3. Optimization of reaction conditions for synthesis of α-mono-bromomethyl ketonesa
表3. α-一溴甲基酮类化合物的合成反应条件优化a
aReaction conditions: 120℃, 1a (0.1 mmol), bromine water solution (Eq. (Br2):(Eq. H2O) = 0.3:14), solvent (1 mL).bIsolated yield.
3.2.2. 底物的扩展
在上述最优条件下,继续研究了该法的底物范围和官能团耐受性(表4)。值得一提的是各种取代底物在脱二溴反应中具有良好的相容性,通过实验观察到芳香环上带有吸电子取代基和给电子基团的α-多卤代物均适用于该方法,获得中等至良好产率的脱溴产物3a~3l,同时发现在α-一溴甲基酮的合成中,缺电子芳香底物、大共轭芳香族底物和脂肪族取代的α,α,α-三溴甲基酮都可以顺利进行,以中等产率得到相应的产物3m~3o。
3.3. 反应机理
为深入地了解反应机理,以α,α,α-三溴甲基4-叔丁基苯基酮为原料,在160℃温度下,以四氢呋喃为溶剂,40 mg (Br2(6.4 mg)、D2O (33.6 mg)) Br2/D2O为催化剂进行了对照实验,获得收率70%的氘化产物,氘代率99%,表明H原子来自H2O (图4)。
Table 4. Synthesis of the α-monobromomethyl ketones 3a~3oa,b
表4. α-一溴甲基酮3a~3o的合成a,b
aReaction conditions: 120℃, 1a (0.1 mmol), bromine water solution (Eq. (Br2):(Eq. H2O) = 0.3:14), solvent (1 mL).bIsolated yield.
基于上述结果和文献报道 [8],提出了可能的反应机理(图5),溴单质和水反应产生HBr和HBrO,HBrO也可以进行歧化反应生成HBr和HBrO3,α,α,α-三溴甲基酮1a和HBr在溶剂1,4-二氧六环等作用下生成中间体A,A继续通过消除反应产生烯醇化的中间体B,最后中间体B异构化获得产物2a,在体系中2a可进一步进行脱溴反应得到3a。
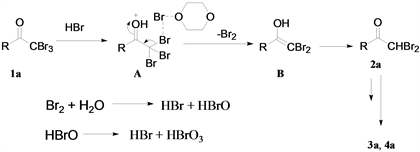
Figure 5. Possible selective debromination mechanism of α,α,α-tribromomethyl ketone
图5. α,α,α-三溴甲基酮的可能脱溴反应机理
4. 结论
本文报道了以1,4-二氧六环为溶剂,通过调节溴水溶液的质量和反应温度,实现了α,α,α-三溴甲基酮的高选择性还原脱溴。以中等至良好的收率合成了α,α-二溴甲基酮和α-一溴甲基酮。是一种基于溴水溶液催化还原的α,α,α-三溴甲基酮类化合物的高选择性脱溴反应,通过调整溴水溶液质量、改变反应温度和溶剂,建立了α,α-二溴甲基酮和α-一溴甲基酮类化合物的合成新方法,该法具有催化剂易得、条件便于控制,官能团耐受性好、底物范围广等优点。通过对照实验,确定了脱溴还原的氢源来自水中的氢,并提出了该类脱溴反应可能的反应机理。
基金项目
新疆维吾尔自治区高校科研计划自然科学重点项目(No. XJEDU2020I015)资助项目。
NOTES
*通讯作者。